
Autoimmune disease is an “invisible epidemic.” These disorders affect at least as many Americans as cancer and heart disease. Despite this, immune-mediated conditions remain under-recognized, under-researched, under-diagnosed and under-treated. Therefore, we at Your Autoimmunity Connection want to increase awareness and connect patients, families and caregivers with useful resources. So, we publish a series of “Spotlights” on autoimmune diseases and other conditions linked to immune dysfunction. This post shines a spotlight on Myasthenia Gravis (MG).
Myasthenia Gravis
The trigger for this post on Myasthenia Gravis was a voice message from a cherished friend after many years. I was shocked to hear his voice. Even in a short voicemail, the vocal effects of the disease are striking. My friend had been suffering from late-onset MG for years. He was recovering from a myasthenic crisis that required weeks in induced coma. So, Paul, this one is for you.
A Note to Caregivers
The primary symptoms of MG are muscle weakness, sometimes crippling. Therefore, MG patients rely on caregivers to get them through flares. Educating and supporting caregivers, who are usually spouses, relatives or friends without medical training or experience, is a major unmet need in MG. Therefore, this post, the links and further reading are intended to help caregivers, too.
What is Myasthenia Gravis (MG)?
MG is a group of rare (prevalence 14-20:100,000 in US, although probably higher[i]) acquired autoimmune diseases. There are even rarer, non-autoimmune, congenital myasthenic syndromes (CMS). We will not discuss those here, but see this link to information about them in the endnotes.[ii]
The name was bestowed on the disease in the late 19th C. However, the first description was in 1672. Even earlier are historical accounts of the illness of Chief Opechankanough, who died in 1644, that strongly suggest he suffered from late-onset MG.[iii]
Myasthenia Gravis is one of those Greek-Latin mashups so common in medical lingo. It means “grave muscle weakness” and that sure sounds awful. MG can be life-threatening (see myasthenic crisis below). Even so, available medical treatment can now give most patients normal life expectancy and good quality of life.
Symptoms and Pathology
Characteristic symptoms include waxing and waning muscle weakness. A hallmark is fatigability, where muscles lose strength with use and recover somewhat with rest. Ocular MG, marked by eye muscle weakness such as ptosis (droopy eyelid), is the most frequent form of the disease.
Bulbar muscle weakness affects the face, jaws, throat and neck. The bulbar nerves (cranial IX-XII) control speech, swallowing, breathing and neck muscles (see illustration). Less often, peripheral muscles are affected, generally upper body and more proximal (shoulders) than distal (hands).
All the same, some of these symptoms occur in other neurological diseases that are more common than MG. This means MG is often misdiagnosed, especially in older men.
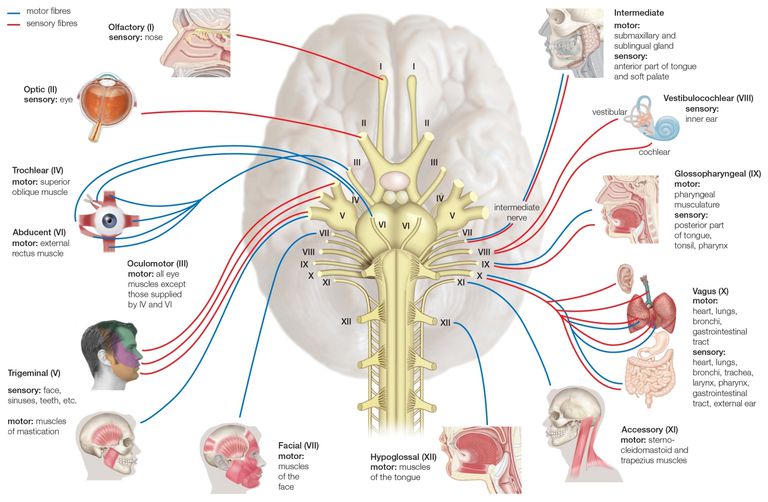
The ocular motor nerves are III, IV, VI, shown in mid-upper left.
The bulbar nerves are IX–XII, described on the lower right.[iv]
What causes MG?
What do all acquired autoimmune MG cases have in common? The answer is going to get a bit technical. The immediate cause is a misdirected immune (T- and B-cell) attack on the neuromuscular junction (NMJ) receptors of skeletal muscle. These are mostly nicotinic (yes, that nicotine) acetylcholine receptors (nAChR) (see illustration).
Since acetylcholine (ACh) transmits motor nerve signals to muscles, damaged receptors produce painless weakness in affected muscles. This variety is called seropositive MG because patients have detectable antibodies to nAChR in their blood.
Seronegative (no detected nAChR antibodies) patients have different targets. Sometimes those targets are Muscle-Specific Kinase (MuSK) receptors, which also mediate ACh signaling. A third receptor target is titin, a striated muscle antigen. The latter is generally not found in early-onset MG, but seen in 50% of late-onset patients.[v]
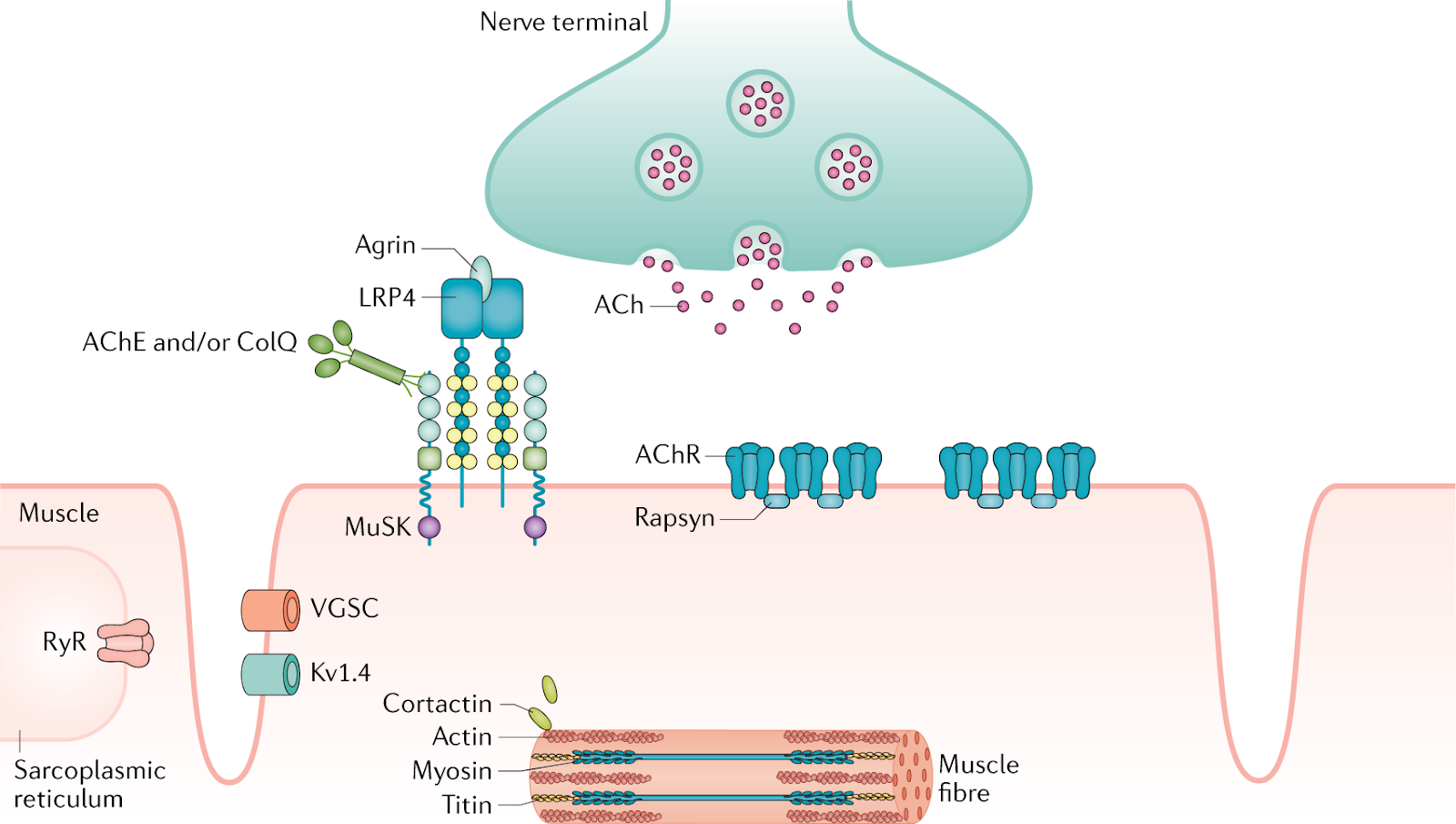
Figure 2. Schematic of striated muscle nerve junction (NMJ). Shows the ACh neurotransmitter released by nerve and binding to AChR sites on muscle cells. Also shown is the MuSK receptor and deeper in the muscle, the titin receptors. All are potential targets of damage in MG.[vi]
The role of the thymus
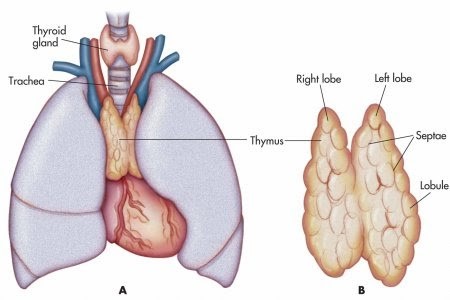
between the lungs and ventral to the heart.
The thymus gland is where immune system T-cells mature and learn to differentiate self from foreign antigens. It gives T-cells their name.
The thymus is abnormal in some 75% of MG patients. Thymic hyperplasia (enlargement) is common. This is similar to thyroid hyperplasia in Hashimoto thyroiditis. In about 10% of patients, thymomas, tumors in the thymus gland, trigger MG. These patients especially, but also some patients without thymomas, benefit from thymectomy, surgery to remove the thymus.
Types of Myasthenia Gravis
For such a rare disease, MG comes in a surprising variety of forms. MG is classified by age of onset. However, note that researchers use several different arbitrary cutoff ages for early- and late-onset MG.
- Transient neonatal MG: mothers with MG may transfer anti-nAChR antibodies to their newborns. With supportive care, most of these babies recover completely within a few months.
- Pediatric or Juvenile onset MG: Less than 10% of cases. Like type 1 diabetes, a childhood autoimmune reaction to unknown triggers.
- Early-onset MG: primarily (3-4:1) females aged 20-30. This used to be the more common form, but as the population lives longer, more late-onset disease is being detected.
- Late-onset MG (LOMG): primarily males aged 50-60, sex ratio equal after 70. Until recently, LOMG in men was under-detected, but evidence is increasing that actual incidence has grown, too. [vii]
Across cases at any age there are five classes of severity (Osserman classification)[viii]:
- Ocular weakness
- Mild weakness:
- Limbs
- Bulbar
- Moderate weakness (limbs, bulbar)
- Severe weakness (same subtypes)
- Intubation or ventilation needed (myasthenic crisis)
A myasthenic crisis is a severe, life-threatening episode of weakness. If it affects swallowing or breathing the patient may require intensive care, intubation, ventilation, even induced coma.
Triggers of MG
Like most autoimmune diseases, symptoms wax and wane, with flares often set off by specific triggers, such as:
- Infections, especially viral infections
- Vaccinations (although MG patients should get vaccinated against flu and other viral diseases)
- Anesthesia
- Certain drugs, especially antibiotics and cardiovascular
- Emotional stress
- Menstruation
- Temperature extremes
- Bright sunlight may trigger ocular symptoms
A family history of MG is unusual. However, family members with other autoimmune diseases (e.g., MS, RA) are more frequent and suggests a genetic predisposition. Nevertheless, the genomic patterns are poorly understood. Moreover, MG patients are more likely to have other autoimmune diseases. Especially likely are Hashimoto thyroiditis, rheumatoid arthritis, scleroderma, lupus, and Sjogren syndrome.
How is Myasthenia Gravis Diagnosed?
As with other autoimmune diseases, diagnosis is often difficult or incorrect. This is especially true for late-onset disease in males. Many practitioners often do not even consider MG in the differential diagnosis.
This is particularly true of ocular MG (OMG). Ophthalmologists are often the first physicians consulted for eye weakness. However, MG is usually not high on their index of suspicion. For eye weakness there are several alternative diagnoses, from blepharospasm and cranial nerve palsies to stroke.
In patients with bulbar or general symptoms, there are also many alternative diagnoses. These include ALS, botulism, brainstem gliomas (tumors), Lambert-Eaton Myasthenic Syndrome, Multiple Sclerosis (MS), progressive bulbar palsy, thyroid disease, stroke and others.
Physical and laboratory tests for MG
Legacies of old school medicine, physicians still use a few hands-on clinical tests, including resting eyelid and ice-pack tests. Imaging frequently shows an enlarged thymus. Electrophysiologic nerve stimulation (ENS) testing can detect abnormal neuromuscular transmission. Pharmacological tests using short-acting acetylcholinesterase inhibitors temporarily increase ACh levels and produce a brief but diagnostic improvement of symptoms. More recently, immunologic testing can detect elevated AChR, MuSK or titin antibodies in blood. Unfortunately, these are costly tests only used when the index of suspicion for MG is high. Thyroid testing may also be worthwhile, since 4-5% of MG patients have concurrent autoimmune thyroid disease.[ix]
There is an unmet need for faster and more accurate detection and diagnosis. Early detection is key since patients respond best to early treatment. As with other rare diseases, education, awareness, analytics and decision-support technology may help medical practitioners consider MG more frequently. Hopefully, such approaches can move patients up the care pathway to more specific diagnostic tests.
How is MG treated?
Once diagnosed, there are four major modalities for MG treatment. These are: surgery, acetylcholinesterase inhibitors, immunosuppressants (corticosteroids, etc.). For myasthenic crisis, there are rescue treatments, including plasmapheresis and intravenous immunoglobulin.[x]
Surgery
Carl Weigert in 1901 was the first to describe a myasthenic patient with a thymic mass. Once the role of the thymus was suspected in MG, surgery became a treatment option. Thymectomy provides robust remission in 80% of patients with thymoma. Some patients with other thymus abnormalities also benefit dramatically from surgery. Ocular MG-only patients are the least likely to benefit from thymus surgery.
Pharmaceutical treatments
Pharmacologic treatments for MG are complex. Most patients require two classes of drug to achieve remission. Many then need additional drugs to cope with the side effects of the main regimen!
AChE Inhibitors
Before the 1930s, there were no pharmacological treatments for MG, and mortality rates were high. In 1934, Mary Broadfoot Walker published a discovery based on her experience treating curare paralysis. She showed that injections of physostigmine, an acetylcholinesterase inhibitor, relieved MG symptoms. This lead to her insight that MG was a disease of the NMJ. Physostigmine has particularly harsh side effects. Therefore, it has been largely replaced by pyridostigmine bromide (Mestinon, patented 1945, and generics). Some 60% of MG patients receive pyridostigmine bromide. Some receive other drugs in the AChE inhibitor family such as neostigmine.
Dosing of AChE inhibitors can be tricky. Unfortunately, overdoses resemble some of the symptoms of the disease itself. This means patients and practitioners must monitor dose response and keep the effective dose as low as possible.
Nevertheless, oral acetylcholinesterase inhibitors remain first-line treatments for MG. However, these drugs do not stop the underlying autoimmune disease progression. They rarely relieve all symptoms completely, and many patients become refractory. Therefore, patients are usually prescribed concomitant immunosuppressants or other immune system modulators.
Immune suppressants
Corticosteroids were the first immunomodulator drugs to be used in MG. Prednisone, prednisolone and methylprednisolone may be familiar to readers from their widespread use in other autoimmune diseases. They quickly and effectively mute the autoimmune response, but have notorious side effects. These include weight gain, bloating, GI discomfort and higher risk of infection. Moreover, patients need to titrate (reduce doses gradually) in order to quit or change drugs.
Non-steroidal immunosuppressants used to prevent transplant rejection are also used in MG. These include cyclosporine, tacrolimus and methotrexate, among others. Side effects are similar to steroids and differ from patient to patient. Physicians may have to tweak dosing regimens to provide the best primary effect and the most tolerable side-effects.[xi]
Biologicals: Few specialty drugs are specifically approved for MG. Only one monoclonal antibody biologic, Soliris (eculizumab), is approved (since 2017) for generalized MG (gMG).[xii] Off-label use of other MAbs, especially rituximab[xiii] has shown promise in MG and clinical trials are underway. Cladribine, a targeted anti-lymphocyte drug approved for MS, has also shown encouraging results in a small study.[xiv] Several investigational drugs are in various stages of progress.[xv]
Clinical Trials: Quite a few clinical trials are ongoing. See these resources.[xvi] [xvii] [xviii]
Rescue therapies: Immunoglobulin infusions and plasmapheresis
During a myasthenic crisis, emergency supportive care (intubation, ventilation, coma) may be needed. This is obvious in patients with swallowing or breathing difficulties that can become life-threatening within hours. Intensive care may include plasmapheresis or immunoglobulin infusions. Plasmapherisis cleans the patient’s blood through a system similar to that used in kidney dialysis patients, but one that removes AChR antibodies. Immunoglobulin infusions bind and neutralize the antibodies, keeping them from further damaging the ACh receptors.
Other treatment considerations
Side effects are a big issue for MG patients.[xix] All drugs used have GI side effects. Careful timing of doses before meals can mitigate these effects. Even more so, the effect of the drugs may also help with swallowing problems. AChE inhibitors can cause nausea, vomiting and diarrhea. Other side effects include muscle cramps, weight loss, headache, insomnia and abnormal dreams. Corticosteroids are notorious for appetite stimulation, weight gain and bloating, and all immunosuppressants for increasing risk of infection and lymphomas. Complicating things further is dose sensitivity, which varies by patient and over time and must be carefully managed. Most MG patients are on at least two drugs. Many are taking more than two, making disease management more complicated for patients and their caregivers.
Particularly important for MG patients is a long list of drugs to avoid.[xx] These include common antibiotics, especially the aminoglycosides and quinolones, neuromuscular blocking agents, opioids and other CNS inhibitors, anticonvulsants, and others.
Anesthesia and vaccination present particular challenges to MG patients. General anesthesia generally exacerbates symptoms, and the anesthesiologist has to be especially attentive to these effects. Since viral infections can exacerbate weakness, MG patients should receive influenza and other viral vaccines, even though the vaccines themselves may provoke flares. Careful monitoring of MG patients after vaccination is therefore necessary.
Beneficial lifestyle changes include avoiding excessive heat, reducing stress and eating a high-potassium diet. Patients, caretakers and practitioners need to watch out for potential flares triggered by other drugs.
Written by: Ellen M. Martin
For further reading
(all the links in the notes contain a wealth of further information, too!)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1626141/#B4
https://rarediseases.org/rare-diseases/myasthenia-gravis/
Sources
[i] https://myasthenia.org/For-Professionals/Clinical-Overview-of-MG
[ii] https://www.mda.org/disease/congenital-myasthenic-syndromes
[iii] https://jamanetwork.com/journals/jamaneurology/article-abstract/587278
[iv] https://www.thoughtco.com/cranial-nerves-function-373179
[v] https://jamanetwork.com/journals/jamaneurology/fullarticle/774608
[vi] https://www.nature.com/articles/s41572-019-0079-y
[vii] https://jamanetwork.com/journals/jamaneurology/fullarticle/774608
[viii] https://www.slideshare.net/smcmedicinedept/myasthenia-gravis-pathophysiology-cl-features-dd
[ix] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4278125/
[x] https://my.clevelandclinic.org/health/diseases/17252-myasthenia-gravis-mg-/management-and-treatment
[xi] https://www.mda.org/disease/myasthenia-gravis/medical-management
[xiii] https://myastheniagravisnews.com/rituximab-for-myasthenia-gravis/
[xv] https://myastheniagravisnews.com/experimental-treatments-for-myasthenia-gravis/
[xvi] https://myasthenia.org/Research/Clinical-Trials
[xvii] https://www.centerwatch.com/clinical-trials/listings/condition/674/myasthenia-gravis-generalised/
[xviii] https://www.clinicaltrialsregister.eu/ctr-search/search?query=Myasthenia+Gravis
[xx] https://www.mda.org/disease/myasthenia-gravis/medical-management




3 Comments
lordmyrt · June 9, 2020 at 5:48 pm
Today, June 6, is the anniversary of the birth of Henry Hallett Dale, who discovered Acetylcholine.
https://www.sciencedirect.com/science/article/pii/S1631069106000485
lupita_brasil · November 17, 2020 at 1:21 am
I am very glad I came across this post and stopped to read it from beginning to end, as it left on a very positive note. I was in the beginning of my own journey with MS and the depression it’s was giving me was unbearable , I found some encouragement from several blogs and last year in seeing Rochelle make her personal goals after overcoming the disease with natural medicine I have to tried it also .I’ve kind of resigned to the fact that this is how life will be for me back until I found herbs that stop this multiple sclerosis easily and relief all the Fatigue and other symptoms I was experiencing ,I’m passing this info to anyone at there because ww w .multivitamincare .org has the right cure and caregiver to this disease ….I took various supplements, medicine prescribed by neurologist,massage and physiotherapy still the disease is was progressing very fast until the the MS formula from that caregiver .
Post-Viral Syndromes - A History of Neglect (Long COVID Part 2) – Your Autoimmunity Connection · January 10, 2021 at 5:53 pm
[…] We also know that many autoimmune conditions are highly correlated with viral infections. For example, symptomatic Epstein-Barr Virus (EBV) infection, aka mononucleosis, may trigger several autoimmune diseases. The list includes cases of multiple sclerosis (MS) and systemic lupus erythematosus (SLE) aka lupus. Researchers have proposed additional viral and bacterial infections as triggers of SLE. More recently, research has identified human herpesvirus 6 (HHV6) as a risk factor for MS. The association of rheumatoid arthritis (RA) with periodontal disease likely follows bacterial, rather than viral infections. However, RA also follows Epstein-Barr and cytomegalovirus infection. Viral infections may trigger autoimmune flares as well, e.g., myasthenia gravis. […]